
A further reduction on execution time is possible by parallelization. Here, however - especially for adaptive tree-type methods - the implementation is quite difficult and cumbersome.
Our approach, which we have now implemented, is a variant of the adaptive Barnes-Hut/Multipole method (see also: J.K. Salmon & M.S. Warren, Int. J. Supercomp. App. , Vol.8.2). We use a hash-technique for dealing with the adaptivity of the method and parallelize with space-filling curves by assigning segments of the increasingly ordered hashtable-key list to each processor.
Altogether this results in an efficient long-range (Coulomb, van der Waals) force evaluation without potential cut-off and a simple incorporation of short-range forces.
We have applied this approach to the simulation of a NaCl-melting process and use it also in the examination of the dynamical behaviour of the Bovine-Pancreatic-Trypsin-Inhibitor (BPTI).
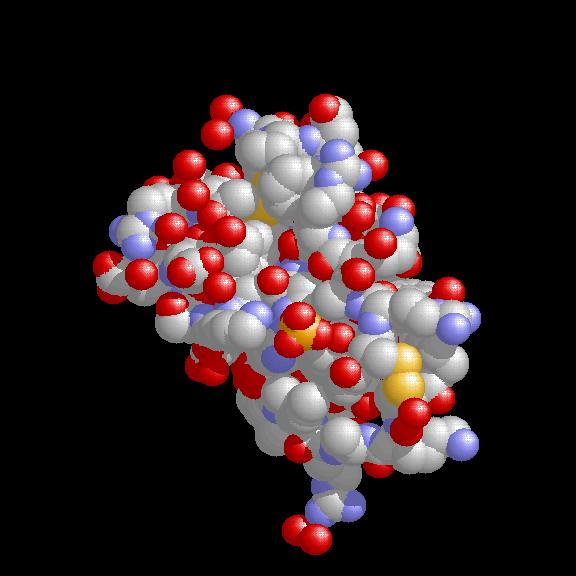

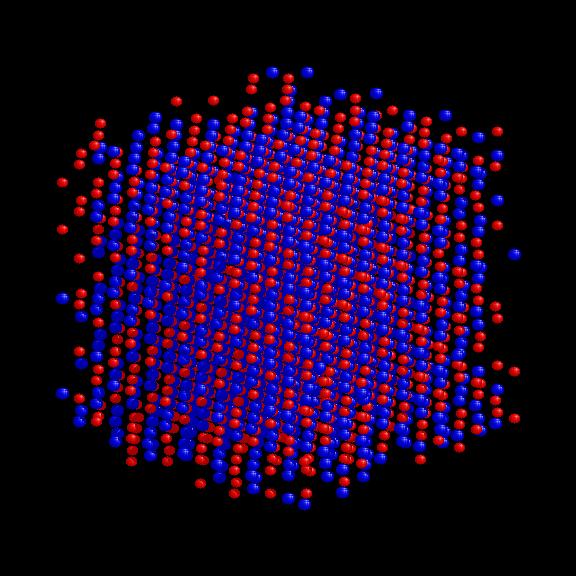


Startconfig.: uniform grid, 1014 Na, 1014 Cl ions, Maxwell-Boltzmann velocity distribution at 1800 K. Particle configuration for NaCl at time 0.47, 0.94 and 1.41 (time-unit 1.03e-15 sec.).